Research Interests
P. Schienbein. J. Chem. Theory Comput. 2026
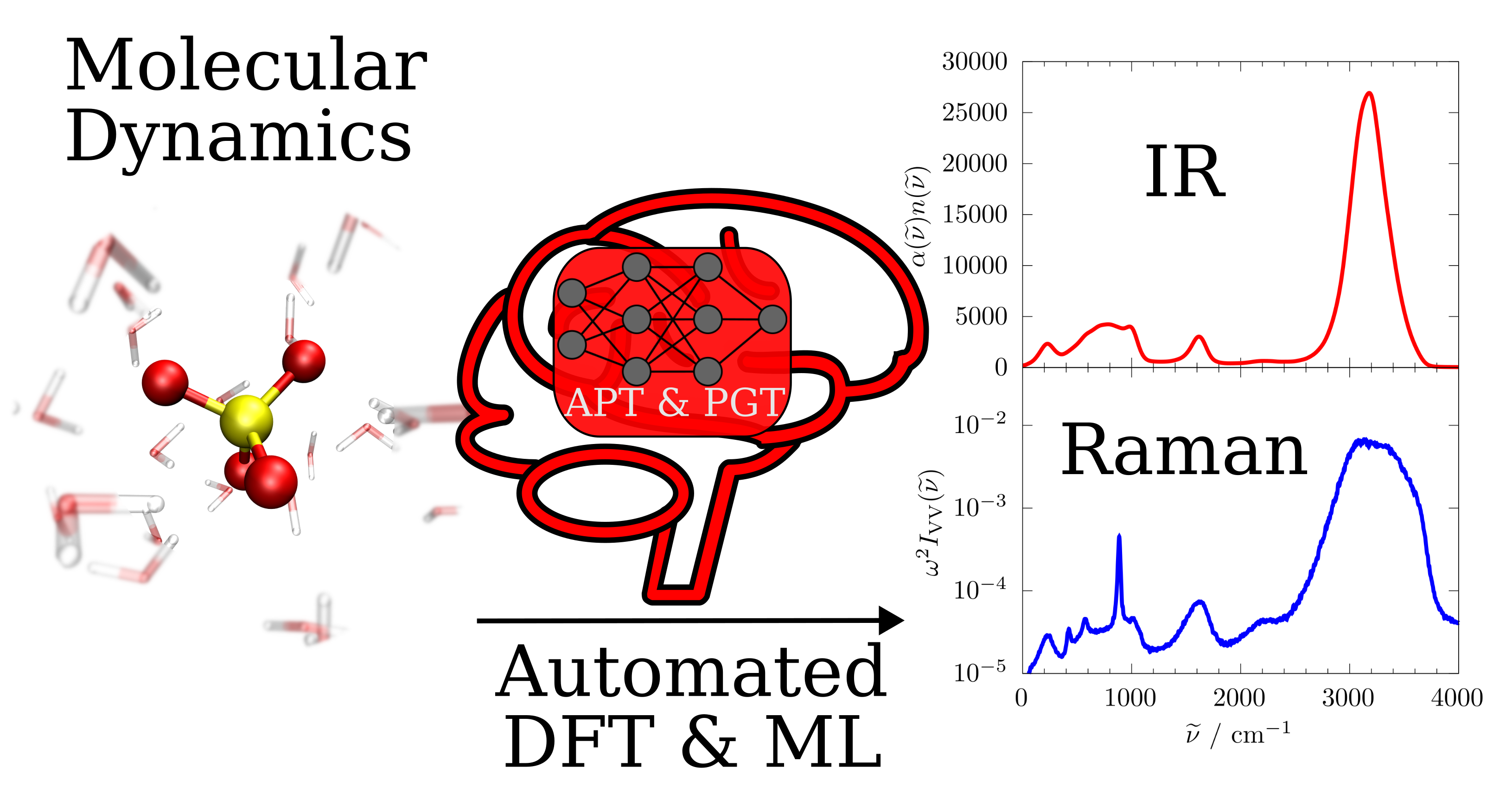
Predictive Vibrational Spectroscopy
Vibrational spectroscopy provides a uniquely sensitive window into the structural dynamics of atoms and molecules in condensed phases, revealing how intermolecular interactions and local environments shape molecular behavior [Chem. Rev., 2026]. Our research focuses on developing predictive, atomistically resolved vibrational spectra from machine-learning molecular dynamics (MLMD) simulations with full electronic-structure fidelity. Calculating such spectra requires sufficiently long trajectories and large enough system sizes to capture thermal fluctuations, intermolecular dynamics, and collective modes in condensed phases. Machine learning makes these simulations feasible while retaining the essential physical ingredients of first-principles methods, including anharmonicity, finite-temperature dynamics, and explicit electronic effects. This level of accuracy is essential not only for reproducing experimental spectra, but for interpreting them in terms of the underlying molecular dynamics and intermolecular interactions. Explicit electronic polarization is particularly critical for systems such as liquid water, where important spectroscopic signatures — including hydrogen-bond modes — arise from collective electronic responses and are therefore absent in non-polarizable force fields, limiting a microscopic interpretation of the observed dynamics.
To enable predictive infrared and Raman spectroscopy from MLMD trajectories, we develop machine-learning models for spectroscopic response properties, including atomic polar tensors (APTs) and polarizability gradient tensors (PGTs). Recently, we introduced machine learned APTs [J. Chem. Theory Comput., 2023] and PGTs [J. Chem. Theory Comput., 2026], providing a rigorous framework for the efficient calculation of anharmonic IR and Raman spectra in complex molecular systems. The corresponding neural network implementations are available on [Github] within the Mimyria software framework.
Time-Resolved and Nonlinear Spectroscopy
Beyond equilibrium spectroscopy, we are interested in simulating time-dependent and nonlinear spectroscopic observables, including pump–probe spectroscopy in condensed phases [J. Phys. Chem. Lett., 2025]. These approaches provide direct insight into ultrafast structural rearrangements, vibrational energy transfer, and nonequilibrium molecular dynamics at finite temperature. By combining MLMD simulations including external electric fields [Nat. Commun., 2024] with predictive spectroscopic observables [J. Chem. Theory Comput., 2026], we can simulate fully anharmonic, temperature-dependent pump–probe signals with electronic-structure accuracy. Importantly, the atomistic nature of the simulations enables us to directly connect transient spectral features to the underlying molecular motions and intermolecular rearrangements, allowing spectroscopic experiments to be interpreted in terms of microscopic dynamics rather than phenomenological models alone.
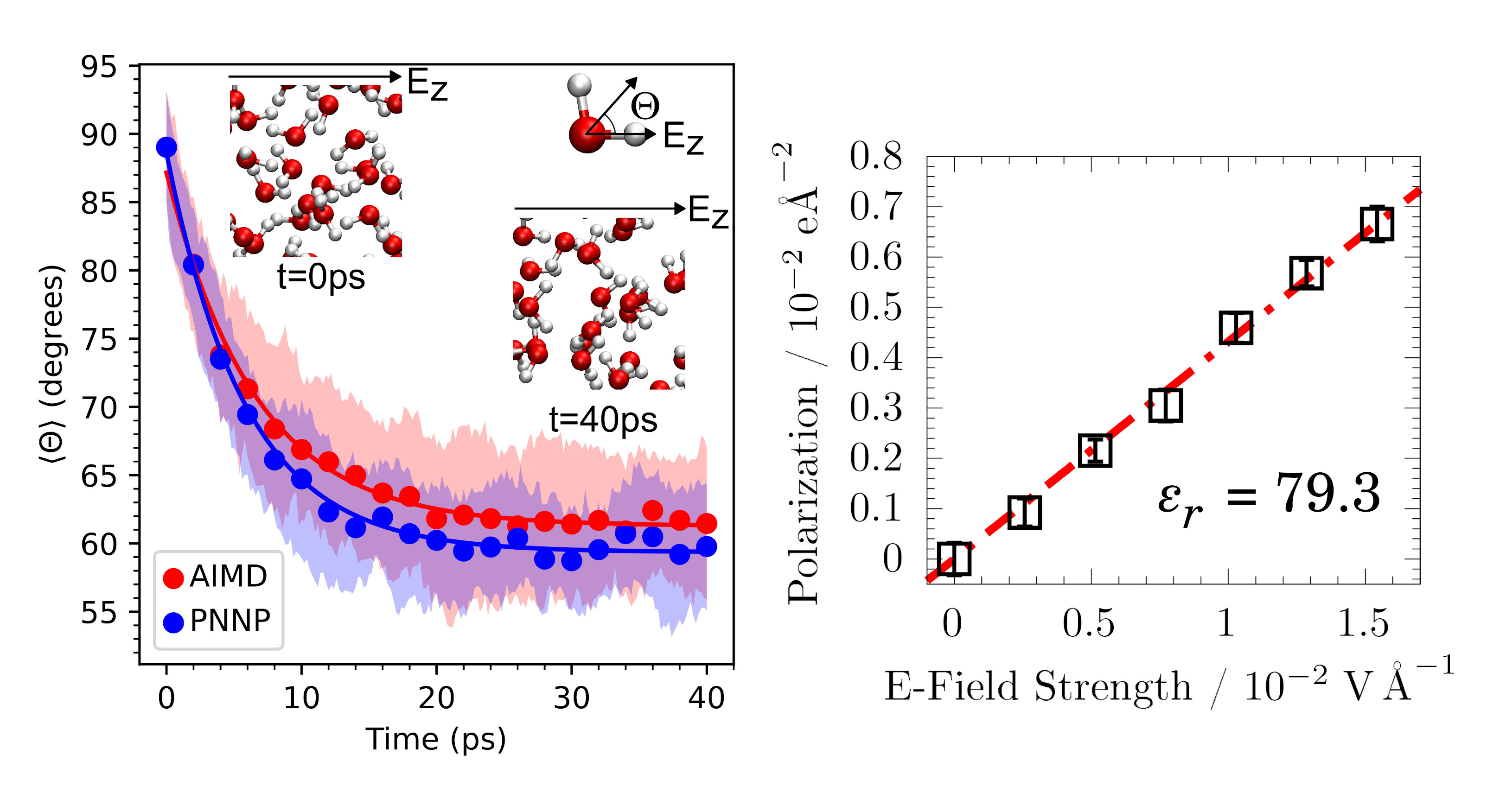
Electric Field Simulations
Electric fields are omnipresent in nature and technology. They play a central role in a myriad of electronic and energy conversion devices, including field effect transistors, (super-)capacitors, batteries and solar cells. In chemistry, electric fields can be used to steer selectivities in catalysis and to control reactivities. Finally, they are present at electrified interfaces, which are currently a major research topic in theoretical chemistry. We have recently introduced a perturbative approach to accurately treat external electric fields in machine learning molecular simulations based on the atomic polar tensor [Nat. Commun., 2024]. The code to conduct such simulations is available on [Github]. Being able to conduct those simulations is a major ingredient of our current research, for instance to calculate conductivities in electrolytes which are of crucial importance in battery development. We have recently employed the same machinery to model a THz light pulse interacting with liquid water [J. Phys. Chem. Lett., 2025].
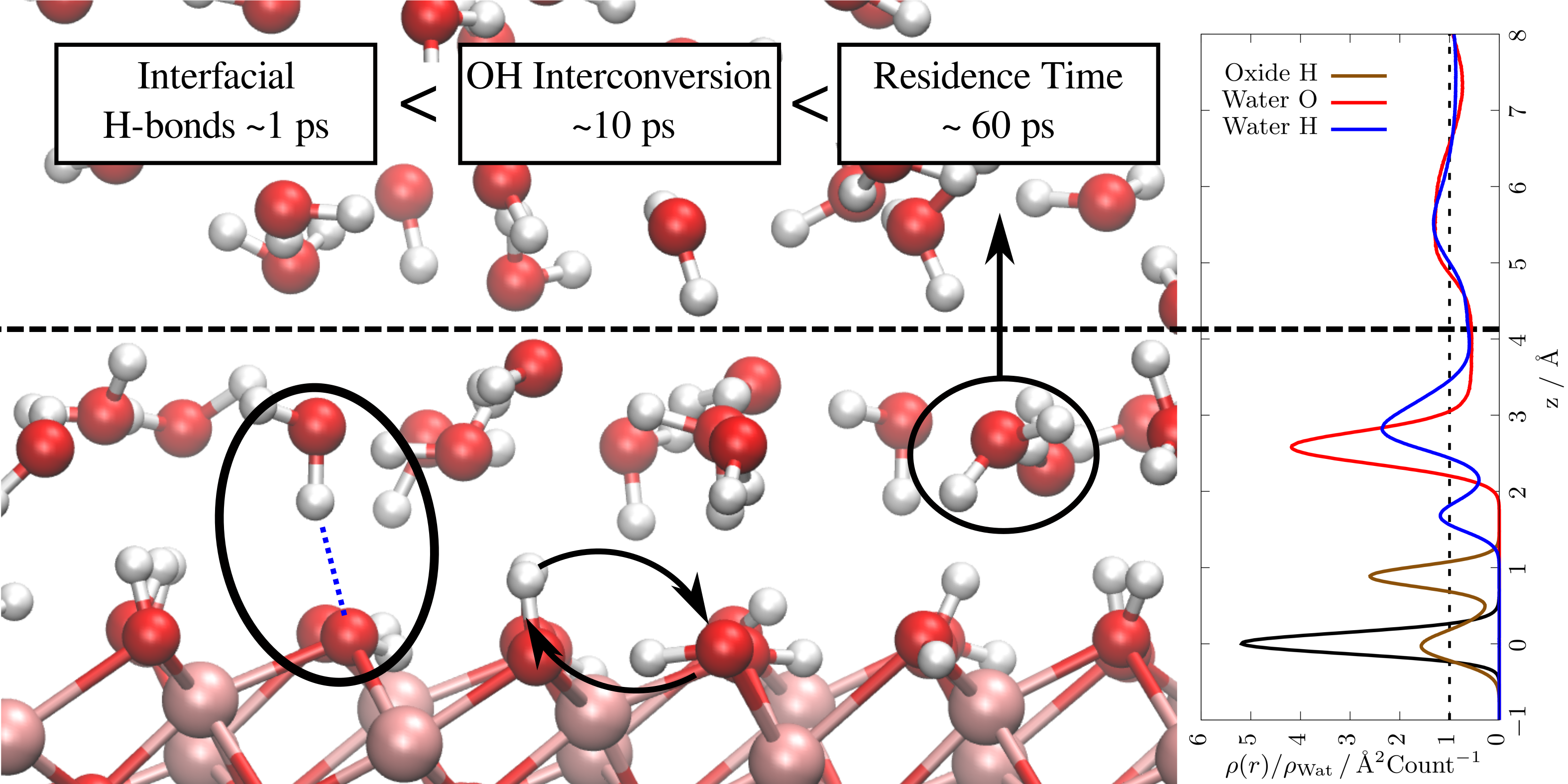
P. Schienbein and J. Blumberger. Phys. Chem. Chem. Phys. 2022, 24, 15365
Metal Oxide / Liquid Interfaces
Metal oxide/water interfaces play an important role in biology, catalysis, energy storage, photocatalytic water splitting, or fuels from CO2 (artificial photosynthesis). Our research focuses on using molecular simulations to uncover the solvation dynamics at such interfaces, for example of the hematite/water interface [Phys. Chem. Chem. Phys., 2022]. Machine learning potentials have been an indispensable tool to accelerate these simulations, thereby allowing us to converge dynamical properties which is not possible using brute-force ab initio molecular dynamics. Another complementary aspect are thermodynamic properties, particularly free energy differences which determine the microscopic structure. One example are protonation states which are governed by their corresponding deprotonation free energies. Recently, we have devised a systematic workflow to calculate such free energies using thermodynamic integration [ChemPhysChem, 2024] or Umbrella sampling [J. Phys. Chem. Lett, 2025] from machine learning potentials and active learning.
Liquids and Solutions
Liquids and solutions are essential to daily life, particularly liquid water (“the matrix of life”1) and aqueous solutions. Understanding solvent-solute interactions is crucial, as solvents actively influence chemical reactions, thermodynamic properties (e.g., melting and boiling points), and conductivities. These interactions also affect protein folding in biological systems and determine the bioavailability of pharmaceuticals. Previously, we studied water along its liquid-vapor phase diagram up to the supercritical phase, demonstrating that the characteristic hydrogen bond network of liquid water largely dissipates under these conditions [Angew. Chem. Int. Ed., 2020] [Science Advances, 2025]. Employing our approach to perform electric field simulations (see above), we could recently compute conductivities of aqueous salt solutions and explore the transition from vehicular to structural motion of ions as charge carriers in liquid water [J. Phys. Chem. B, 2026].